BiSearch Web Server User's Manual
Contents
Simple tasks
Calculating melting temperature of a primer
This task can be reached by clicking the 'Primer tm' link in the left-side menu. Users should provide only the sequence of their short oligo nucleotide. By clicking the 'Calculate' button the server calculates the melting temperature of the oligonucleotide by using a modified neighbourhood method.
Melting temperature in Celsius is calculated by the following formula:
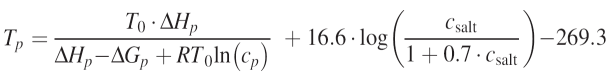
where T0 = 298.2 K, R = 1.987 (cal/mol K) the molar gas constant, DHp and DGp are the enthalpy and free energy of the primer p, p=(p1, . . . , pn) which are computed according to the nearest neighbor schemes, i.e.
DHp=-1000*[2*DHe+Sum(i=1->n-1)(DH(pi,pi+1)] and
DGp=-1000*[2*DGe+DGi+Sum(i=1->n-1)(DG(pi,pi+1)],
where enthalpy and free energy of a string consisting of two bases is given in Table below and DHe = 5 kcal/mol, DGe = 1 kcal/mol and DGi = -2.2 kcal/mol. The dimensionless concentration of the primer in the PCR mixture is Cp and Csalt is the molar salt concentration.-DH(pi,pi+1) -DG(pi,pi+1) AA or TT 9.1 1.55 AT 8.6 1.25 TA 6.0 0.85 CA or TG 5.8 1.15 GT or AC 6.5 1.40 CT or AG 7.8 1.45 GA or TC 5.6 1.15 CG 11.9 3.05 GC 11.1 2.70 GG or CC 11.0 2.3 Calculating the score of a primer pair
This task can be reached by clicking the 'Primer score' link in the left-side menu. Users should provide two sequences, i.e. the sequences of their forward and reverse primers. Click the 'Calculate' button to obtain the score of the primer pair. This score is the weighted sum of the differences of melting temperatures from a predefined value (see Parameters section), percent of GC content, self-annealing and self-end-annealing of the primers as well as pair annealing and pair-end-annealing. The exact form of the weighted sum is described in our manuscript.
Primer-design
General information for primer-design
This task can be reached by clicking the 'Primer design' link in the left-side menu. Users should enter in a plain text format the sequence to be used as a template for primer-design. Users should also enter nucleotide positions to limit the region to use for both forward and reverse primer search and set the maximal PCR product length accepted. It is obligatory to set these parameters before starting primer-design. Other search parameters can be set as well, after clicking on the "Set parameters" button (please see at the appropriate section).
Primer-design for bisulfite treated DNA
If user wishes to design primers for bisulfite treated DNA template the "Bisulfite treatment" option should be selected. After the bisulfite treatment the complementarity of the two original DNA strands is lost. Primer pairs should be designed to amplify either the treated sense or antisense chain. The sofware proposes both options, user has to mark his/her preference. By default, the software uses the sense strand.
The template sequence should be entered in a plain text format, WHITHOUT BISULFITE CONVERSION. The software converts automatically the sequence if the "Bisulfite treatment" option was selected. The converted sequence does not appear on the screen. The software memorizes the original CpG sites and uses this information for the calculations. This would be impossible if a bisulfite treated sequence is entered.
User should also determine the maximum acceptable Tm difference between the annealing of a primer to the methylated and unmethylated forms of the sequence. If this value is different from 0, the software may propose a primer with CpG dinucleotide(s) in the sequence. In this case alternative nucleotides are indicated at these specific positions (i.e. Y - C or T for the sense chain and R - A or G for the antisense chain). The default maximal temperature difference is 2.5 C.
Primer-design for methylation specific PCR
When using MSP the objective is the selective amplification of methylated target molecules. The software asks the minimal acceptable Tm difference between the annealing of a primer to the methylated and unmethylated forms of the sequence. The default value is 8 C. For further detail on primer-design please refer to the previous section.
Genome search
General information for genom search
Genome search task can be reached by various way. User may search genomes by using a short oligonucleotide sequence, by using a primer pair, or by selecting primers after primer-design.
In each case, search may be carried out on four different genomes, namely: Homo sapiens, Mus musculus, Rattus norvegicus and Pan troglodytes. If user wishes to search on bisulfite treated genomes the "Bisulfite converted genome" option should be selected. If users would like to search on other genomes, please contact BiSearch On-line Assistance.
The position specific mismatches can be entered each case too. This means the total number of mismatches accepted from the 3' most nucleotide to each specific position. This creates a string, where each number corresponds to a specific position in the primer. The first position in this string is the 3' most nucleotide and the last one corresponds to the 5' end of the primer. The software will use this string for the genome search. The hits shown on the results page will not have more mismatches with the entered query sequence than accepted by the string.
Searching user's primer in a genome
This task can be reached by clicking the "Simple Genome Search" link in the left-side menu. Users should provide only the sequence of their short oligonucleotide.
Searching user's primer pair in a genome
This task can be reached by clicking the "Primer Genome Search" link in the left-side menu. After the simple primer search carried out by the software (please refer to the previous section) the genomic locations of the hits are analyzed. If two hits on two complementary strands are found within less than 1 kb the software considers the sequence neighbored by the primer(s) as a PCR product and shows it on the results screen.
Searching after primer-design
The primer-design and search function of the software can be used sequentially. The primer pairs proposed by BiSearch Web Server are shown in a tabular form. Primer pairs can be directly selected from the table and sent for genome search. If a primer pair was designed for a bisulfite treated target sequence the search will be carried out automatically on the bisulfite treated genomes. For further details on the genome search function please refer to the previous sections.
Parameters
General information
The software uses several parameters taken into account for the calculation of primer scores. These parameters have default values but User can modify them. Some parameters, appearing on various screens, were already described in the appropriate sections. Here comes a short description of parameters appearing after clicking on the "Set parameters" link in the left-side menu. In case of simpe parameters like ion and primer concentrations the software demands a unique value. For more complex parameters the software asks for an acceptable range with an optimum value. The distance from the optimum is calculated according to adjustable weights and is included in primer-design. These parameters are the primer length, GC content, Tm and annealing values. The default range for the GC content is acceptable between 40 and 60%, the optimum is 50%. The primer score calculations use a weight of 1 to determine the distance from the optimum. We suggest that the accepted range should be changed when User wishes to design primer pairs for bisulfite treated DNA target. We propose a range of 0 to 60% with an optimal value at 30%.
Adjusting parameters
Parameters can be adjusted after clicking on the "Set parameters" link in the left-side menu. Then the parameters can simply be overwritten. The modified values are taken into account by the server only after clicking "Set parameters" button.
Theory
Undesired PCR products
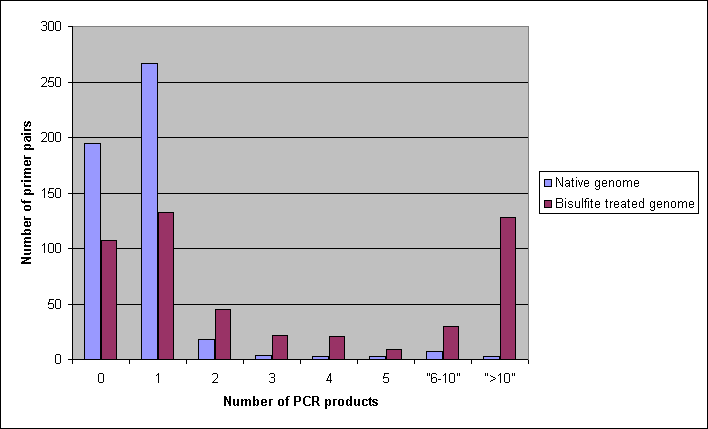
We carried out in silico experiments to demonstrate the differences of amplification of undesired PCR products on native and bisulfite converted genomes. First, 500 primer pairs used in RT-PCR experiments were downloaded from the RTprimerDB (http://medgen.ugent.be/rtprimerdb) and tested for genomic binding sites and PCR products on native and bisulfite treated genomes with the default parameters of BiSearch. Approximately 40% gave no product (i.e. the distance of the primers is more than 1kb), while more than 53% gave a unique product on the native genome. The same dataset analyzed with the same default parameters on the bisulfite treated genome gave a completely different picture. Only 20% gave no PCR product, meaning that half of the primer pairs, which did not amplify on the native genome because of the too big distance of the primers, amplified undesired ePCR products. 25-25% of the primers gave unique and more than 10 amplicons, respectively.
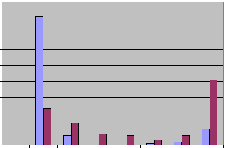
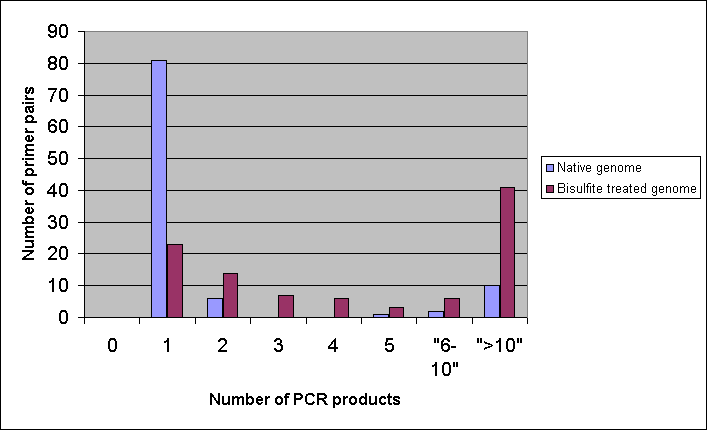
In a second model we generated 100 random genomic primer pairs between 300 and 500 bp distances from each other. The primers were all 20-mers and were in GC-reach regions. First, we used very stringent conditions and only complete matches were considered. This gave similar results for the native and bisulfite treated genomes, with 11 and 15% of multiple ePCR amplicons, respectively. However, when the slightly less stringent default parameters were used the picture became dramatically different. Still more than 80% of the primers amplified a single and only 10% amplified more than 10 amplicons on the native genome. In contrast, according to the prediction, only slightly more than 20% amplified a unique product on the bisulfite treated genome and more than 40% were predicted to amplify more than 10 amplicons. Altogether these results indicate that indeed, the risk of undesired PCR products is much higher on bisulfite converted than in native (genomic) templates.